The efficiency of pharmaceutical R&D is declining. That much is not in doubt: record R&D expenditure in the last decade has led to fewer drug approvals overall, and almost a complete drought in genuine, first-in-class innovation. Set aside biologicals, and the performance in small molecules has been even more dire. But that’s where the unanimity ends. Dozens of hypotheses circulate to explain the decline in efficiency of small molecule drug development – and every part of the process has come under close scrutiny.
DrugBaron has extensively explored the weaknesses in preclinical and clinical development of the lead compound, but largely ignored the foundation of the whole process: designing the right molecule in the first place. It turns out that tweaking medicinal chemistry strategies is just as urgent as tweaking development pathways.
“Applying the Exupéry Principle of design to consumer electronics has turned Apple into the world’s most valuable company. Can it do the same for pharmaceuticals?”
The nub of the problem, it seems, is that pharma companies have overfished the pond. Talking to visionary medicinal chemist Dr David Fox, I was shocked to discover that less than 1% of stable, easily-made small molecules have ever been synthesised, let alone tested for biological activity. The other 99% represent virgin territory waiting for those clever enough to seek them out. If there really are unexplored, unknown continents out there, how do we find them? DrugBaron asked David Fox, and here is what he had to say…
“While economists and business developers are busy analysing trends in drug pipelines and discovery costs, and many pharmaceutical companies are changing their business models, increasingly desperate to understand the factors behind declining R&D productivity, medicinal chemists have also been taking stock and reviewing the kind of molecules they make.
“A designer knows he has achieved perfection not when there is nothing left to add, but when there is nothing left to take away” Antoine de Saint-Exupéry
It is now over a decade since Lipinski’s “Rule of 5” was born (1), and chemists are still discussing its impact, analysing more trends and trying to understand the seemingly growing gap between the number of molecules made and the number of new chemical entities (NCEs) launched as drugs (2). More recently, the American Chemical Society’s Journal of Medicinal Chemistry has reached its 50th anniversary. This birthday has prompted reviews of what chemical reactions medicinal chemists use (3) and what molecular structures they make (4).
What do they say and what can we learn? The most obvious trend is that “small-molecule” drugs have got bigger over time (5). But why is that? Is it the widely held belief that selectivity comes only through complexity? The jury is still out on this one (6). Is it the case that larger molecules can bind to their targets more strongly? Possibly (7). Or is there simply a widespread belief that almost all of the simple molecules have been studied or are unpatentable? We will come back to this one.
Another interesting trend has been unearthed: launched drugs have become ‘flatter’. This means that the proportion of carbons that are flat (sp2-hybridised) rather than have three-dimensional tetrahedral structure (sp3-hybridised) has gone up over the years (8). Again, we have to ask why? This time, the answer lies in the reactions chemists are using. Many excellent reactions are available for linking together flat-carbon containing fragments for library building. Reactions of this type have made such an impact that they won shares in the 2010 Nobel Prize for Chemistry for Heck, Negishi and Suzuki. Indeed, a recent review (3) found that fully 60% of the carbon-carbon bonds formed in the medicinal chemistry departments of three large pharmaceutical companies are palladium-catalysed couplings of this general type. Stop and think about that! More than half of the reactions assembling carbon skeletons involved coupling flat fragments. No wonder drugs are getting ‘flatter’.
Of course, the spread of this chemistry has allowed for huge growth in the number of library molecules that can be easily made. It also facilitates the speedy synthesis of large and complex molecules. But at a cost: an increasingly high proportion of the molecules made look somewhat like one another. Certain areas of molecular space have become incredibly densely populated – while, as we shall see in a moment, others remain almost unexplored.
Small-molecule drug discovery has a big future, but the molecules will be small. Ultra-small.
Are these ubiquitous flat molecules the right ones to be focusing on in any case? It has been pointed out that too many aromatic rings (9), especially non-heterocyclic rings (10), reduce the likelihood of successful development of a drug candidate. Conversely, it has been shown that increasing the three-dimensionality of a molecule (proportion of sp3 carbon atoms) can increase its solubility (11).
Proper three-dimensionality may help in other ways as well: it allows a dramatic increase in complexity compared to flat molecules of the same molecular mass.
How? Consider a simple, flat hexagonal benzene ring with six sites of substitution. There are three ways of arranging two different (non-hydrogen) substituents on this ring, and ten ways of arranging three different substituents. But if we instead start with cyclohexane instead of benzene, each carbon is now tetrahedral rather than flat (as in benzene) and has two possible sites of substitution. There are now 11 ways of arranging two different (non-hydrogen) substituents on this ring, and 110 ways (11 times 10) of arranging three different substituents. This represents a massive increase in complexity with hardly any increase in molecular mass.
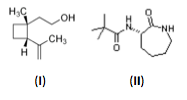 Figure 1: Structure of two ‘ultra-small’ molecules with potent biological activity
Figure 1: Structure of two ‘ultra-small’ molecules with potent biological activity
This is a very simple illustration but the underlying concept shows us that we can generate complexity – and hopefully, therefore, selectivity – in smaller, more soluble molecules. And it works in the real world too: look at the molecules in Figure 1. Here are two examples of compounds that achieve incredible potency and selectivity despite being very small indeed. Grandisol (I), the pheromone of the cotton plant pest Anthonomus grandis (the Boll Weevil) can be detected (by female weevils, anyway) at kilometer distances. (S)-3-(2’,2’-dimethyl-propionylamino)-caprolactam (II) is a sub-nanomolar potency G-protein coupled receptor (GPCR) agonist with impressive anti-inflammatory properties in vitro and in vivo (12). And these are tiny molecules (the caprolactam is 214 Daltons and grandisol only 154 Daltons) with few functional groups. But the groups they do have are clearly presented in the optimal three-dimensional spatial arrangement to give potent binding and hence selectivity.
Simple as this caprolactam looks, at the time of its invention, it was patentable (13). Perhaps more surprisingly, it lies in acres of almost virgin territory in molecular space. According to the online molecular structure generator MOLGEN®, it has over 22 million isomers (14). How many of these isomers have been made as single molecules? According to SciFinder®, just 991 of these isomers have been described (that’s 0.0045%) and only 129 (just over 10% of these) have any biological data associated with them (15).
Of course, it is very likely that many of these 22 million isomers are too unstable or contain essentially impossible functional groups. The caprolactam is a peptide mimetic, with two amides as the only functional groups. So how many molecules of the 22 million isomers have the same arrangement of two secondary amides? According to another program, Assemble 2.0® (16), the answer is 5,453 (but even this does not include possible stereoisomers, so the true answer is much higher). Of these stable, readily made isomers, only 12 (one of which is indeed the caprolactam in Figure 1) have been described at all, and only four have been investigated for biological activity. For sure, more will be known in pharmaceutical company databases, but this powerfully illustrates that even among very small, very simple molecules, the proportion of possible small molecules that have been investigated is very small.
If there are vast tracts of patentable space for “ultra-small” and simple small molecules, what are the advantages of using them? Firstly, they may be more soluble which in turn contributes to the pharmaceutical properties. The caprolactam (II) in Figure 1, for example, has complete oral bioavailability and something approaching ideal pharmacokinetics. The small, less hydrophobic molecules generally have better volumes of distribution, and are less likely to be substrates, inhibitors or inducers of P450 enzymes. By and large, smaller also means easier (and cheaper) to make in bulk, taking the cost of goods out of the equation altogether.
Instead, therefore, of repeatedly shuffling the same deck of largely planar fragments and concatenating them into ever larger units in order to gain binding energy and potency (17), perhaps the future lies in smaller units, but with greater three-dimensional complexity to present a limited portfolio of functional groups in the spatially optimal arrangement.
Too often in the past, the response to discovering a flaw in the properties of a lead compound was to add something to induce a change. More often, molecules grew rather than shrank during lead optimization. But as Antoine de Saint-Exupéry, philosopher, pilot, and author of The Little Prince said: ‘A designer knows he has achieved perfection not when there is nothing left to add, but when there is nothing left to take away.’
FX125L, an analogue of the caprolactam (II) that is in Phase II clinical trials, was subjected to exactly this kind of “Exupéry optimization”: both amide functional groups are absolutely required for activity, and neither the lactam ring nor the alkyl sidechain can be reduced or removed without significant loss of potency.
This “Exupéry Principle” isn’t just about hit-to-lead optimization. The huge strides forward in asymmetric catalysis in the last decade provide routes to enrich molecular libraries with many more “ultra-small” small molecules packed with three-dimensional complexity – in short, to make the composition of a screening library more closely mirror the structural characteristics of biology’s own diverse array of metabolites. Not only could hits from such libraries be closer to developable leads before any optimization takes place, they could show lower cell toxicity, making such libraries more useful for the growing number of functional screens we employ.
Signs are that these principles are already being adopted. A drug as simple as the dimethyl fumarate (Biogen’s BG12) is in late-stage clinical trials. It is a very small molecule yet it, and others like FX125L following behind, are destined for blockbuster status.
The clue is in the name. Small-molecule drug discovery has a big future, but the molecules will be small. Ultra-small.”
Of course, tweaking medicinal chemistry strategies isn’t the sole answer to reversing the decline in pharmaceutical R&D productivity. There isn’t a single answer to the problem. But David Fox makes it very clear there is room for improvement.
Over-fishing, it seems, isn’t just a problem for Atlantic cod. Pharmaceutical companies have spent too long huddled round the same pond. Those that break out further and faster will have the competitive advantage in the next round of the eternal battle for survival and supremacy.
1. (a) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev 1997, 23, 3-25; (b) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev 2001, 46, 3-26.
2. Kola, I.; Landis, J., Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004, 3, 711-715.
3. Roughley, S. D.; Jordan, A. M., The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J Med Chem 2011, 54, 3451-3479.
4. Walters, W. P.; Green, J.; Weiss, J. R.; Murcko, M. A., What Do Medicinal Chemists Actually Make? A 50-Year Retrospective. J Med Chem 2011, 54, 6405-6416.
5. (a) Gleeson, M. P., Generation of a set of simple, interpretable ADMET rules of thumb. J Med Chem 2008, 51, 817-834; (b) Leeson, P. D.; Davis, A. M., Time-related differences in the physical property profiles of oral drugs. J Med Chem 2004, 47, 6338-6348; (c) Lipinski, C. A., Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol 2000, 44, 235-249; (d) Proudfoot, J. R., The evolution of synthetic oral drug properties. Bioorg Med Chem Lett 2005, 15, 1087-1090.
6. (a) Hopkins, A. L.; Mason, J. S.; Overington, J. P., Can we rationally design promiscuous drugs? Curr Opin Struc Biol 2006, 16, 127-136; (b) Schuffenhauer, A.; Brown, N.; Selzer, P.; Ertl, P.; Jacoby, E., Relationships between molecular complexity, biological activity, and structural diversity. J Chem Inf Model 2006, 46, 525-535; (c) Posy, S. L.; Hermsmeier, M. A.; Vaccaro, W.; Ott, K. H.; Todderud, G.; Lippy, J. S.; Trainor, G. L.; Loughney, D. A.; Johnson, S. R., Trends in Kinase Selectivity Insights for Target Class-Focused Library Screening. J Med Chem 2011, 54, 54-66; (d) Huggins, D. J.; Sherman, W.; Tidor, B., Rational Approaches to Improving Selectivity in Drug Design. J Med Chem 2012, 55, 1424-1444.
7. Hajduk, P. J., Fragment-based drug design: How big is too big? J Med Chem 2006, 49, 6972-6976.
8. Leeson, P. D.; St-Gallay, S. A.; Wenlock, M. C., Impact of ion class and time on oral drug molecular properties. Medchemcomm 2011, 2, 91-105.
9. Ritchie, T. J.; Macdonald, S. J. F., The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design? Drug Discov Today 2009, 14, 1011-1020.
10. Ritchie, T. J.; Macdonald, S. J. F.; Young, R. J.; Pickett, S. D., The impact of aromatic ring count on compound developability: further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov Today 2011, 16, 164-171.
11. Lovering, F.; Bikker, J.; Humblet, C., Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J Med Chem 2009, 52, 6752-6756.
12. Fox, D. J.; Reckless, J.; Lingard, H.; Warren, S.; Grainger, D. J., Highly Potent, Orally Available Anti-inflammatory Broad-Spectrum Chemokine Inhibitors. J Med Chem 2009, 52, 3591-3595.
13. Grainger, D. J.; Fox, D. J. Preparation of 3-aminocaprolactam derivatives as anti-inflammatory agents. WO2005053702A2, 2005.
14. Molgen http://molgen.de/?src=documents/molgenonline
15. SciFinder http://www.cas.org/products/scifindr/index.html
16. Badertscher, M.; Korytko, A.; Schulz, K. P.; Madison, M.; Munk, M. E.; Portmann, P.; Junghans, M.; Fontana, P.; Pretsch, E., Assemble 2.0: a structure generator. Chemometr Intell Lab 2000, 51, 73-79.
17. Erlanson, D. A.; McDowell, R. S.; O’Brien, T., Fragment-based drug discovery. J Med Chem 2004, 47, 3463-3482.
18. Rees, D. C.; Congreve, M.; Murray, C. W.; Carr, R., Fragment-based lead discovery. Nat Rev Drug Discov 2004, 3, 660-672.
19. Erlanson, D. A.; Arndt, J. W.; Cancilla, M. T.; Cao, K.; Elling, R. A.; English, N.; Friedman, J.; Hansen, S. K.; Hession, C.; Joseph, I.; Kumaravel, G.; Lee, W. C.; Lind, K. E.; McDowell, R. S.; Miatkowski, K.; Nguyen, C.; Nguyen, T. B.; Park, S.; Pathan, N.; Penny, D. M.; Romanowski, M. J.; Scott, D.; Silvian, L.; Simmons, R. L.; Tangonan, B. T.; Yang, W. J.; Sun, L. H., Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg Med Chem Lett 2011, 21, 3078-3083.
The Cambridge Partnership is the only professional services company in the UK exclusively dedicated to supporting companies in the biotechnology industry. We specialize in providing a “one-stop shop” for accountancy, company secretarial, IP management and admin services. The Cambridge Partnership was founded in 2012 to fill a gap. Running a biotechnology company has little …