Medicines need to be safe and effective.
Once upon a time there was a trade-off between the two that was assessed at the population level – over the entire group of people receiving the medicine was there more benefit than harm. Across a broad swathe of an indication, a positive answer to this risk:benefit equation was usually sufficient to support approval and widespread clinical adoption.
Over the last decade or so, however, this framework has changed substantially, at least for the majority of indications. Increasingly, regulatory authorities and clinicians alike are inclined to consider the risk:benefit equation at the level of the individual rather than the population. Does every individual receiving the medication receive more benefit than harm?
Because of the usual profile of the distribution of risk and benefit over a population treated with a drug, often with a few individuals suffering a disproportionate fraction of the total harm, while the majority gain a lesser benefit, means that the two equations are very different in impact.
“Today medicines have to do more good than harm to every individual treated, rather than to society as a whole”
Consider a drug that has severe side-effects in small number but significant benefits for rest – it doesn’t take much imagination, there are plenty of real-world examples: anti-integrins, such as Raptiva™ and Tysabri™, cause extremely rare re-activation of JC virus leading to PML and death, but offer clinically valuable improvement in the symptoms of debilitating autoimmune conditions; cox-2 inhibitors, such Vioxx™ and Celebrex™ increase the risk of a fatal myocardial infarction in a small number, while offering real clinical benefits in rheumatoid arthritis. It is extremely difficult to weight the balance between a few fatal events and a large and distributed benefit. If you are the individual who bears the negative consequences you are entitled to feel that the risk:benefit equation has been miscalculated, even if there was no way to know ahead of time who would suffer. Increasingly, particularly in the US, the courts have shared this view helping to shape the consensus that drugs must offer more benefit than harm to each individual rather than to society as a whole.
For sure, there are forces pushing in the opposite direction: if you ask patients with a debilitating disease before they start taking a new medicine whether they are prepared to take the risk of a rare, severe side-effect in return for the likelihood of improved quality of life and the answer is invariably ‘yes’. The dissatisfaction with the outcome is usually a post hoc response to the realization that you are the individual who lost the lottery. These positive pressures from patients and patient advocacy groups have resulted in the re-introduction of both anti-integrins and cox2 inhibitors but the safety concerns for the individual continue to dominate the debate over their use, which remains significantly less than it would have been a decade or two ago.
Safety (for decades the elephant in the drug discovery room) has never been a bigger issue than it is today. Billions have been spent on predictive toxicology paradigms designed to identify the merest hint of risk at an early stage in the drug discovery pipeline, before the costs are ratcheted up in later stage clinical development (which remains the only sure-fire way to properly quantify both harm and benefit).
“Safety (always the elephant in the discovery room) has never been a bigger issue than it is today”
The impact of this drive for safety on the productivity of big pharma R&D operations has been well documented, not least in my own recent article. The triple whammy of over-zealous regulators halting development of promising medications on the basis of debatable signs of harm, of drug owners reacting to this by pulling promising product candidates from their pipelines at earlier and earlier stages at the first hint of safety issue, and of clinicians reluctant to prescribe any product that might offer a negative risk:benefit equation to even a tiny fraction of their patients has strangled the drug development industry. We are seeing the greatest retrenchment ever seen, as the behemoths of drug development, who we rely on completely for the next generation of improved medicines, reduce their expenditure on development of novel (and highly risky) new first-in-class medicines in favour of branded generics and other medium-value products that leverage their accumulated expertise in healthcare sales and marketing.
One thoroughly implausible solution to this worrying state of affairs is to hope for a resetting of the public attitude towards risk. A return to a world that remembers the horror of disease unopposed by the modern pharmaceutical industry, a world with child mortality rates that touch almost every household. A populace that recognizes that individually bearing the small risk of a severe negative outcome is a small price to pay for a miraculous cure. While its possible that things (things generally, not just in healthcare) may get sufficiently bad one day in the future that such a “global reset” of attitudes does eventually take place, it is no kind of business model for the healthcare industry to sit and wait for such an event. Where can we look for actions we can take to improve the commercial proposition for developing new first-in-class drug products that really make a material improvement to human health?
“Ironically, the easiest route to improving safety might come from re-evaluating the way we use animal models of efficacy”
Is the solution better predictive toxicology paradigms? Maybe if we could genuinely weed out risky product candidates with better specificity (that is, without throwing away so many promising programmes that are stained by a single concerning finding) things might improve a little. But there is another source of improved safety that might be easier to tap (not least because billions of dollars have already been spent trying to optimize predictive toxicology approaches, so we have to assume the current crop are not far away from being as good as current technology and knowledge allow). Ironically, the easiest route to improving drug safety might come from a re-evaluation of our approach to screening for efficacy in animal models.
Lets consider (in qualitative terms, since a quantitative analysis is impossible) what happens to average harm and average efficacy of all compounds still under consideration as potential products through a fairly typical drug discovery and development pathway.
At the very start of the project all possible molecules (large or small), or at least those that are readily available to the pharmaceutical industry in terms of compound or antibody libraries, represent the field of plausible candidate drugs. Any such compound chosen at random would, if dosed in humans, have a pretty high chance of causing a degree of harm (and for a small molecule library, that harm might be substantial). By contrast, that same randomly chosen compound has an almost zero chance of actually doing something useful. This is illustrated in the figure below: at the beginning of the project (left hand side of the graph) the risk of harm (red line) is high, but the risk of benefit (green line) is low on average for each candidate compound.
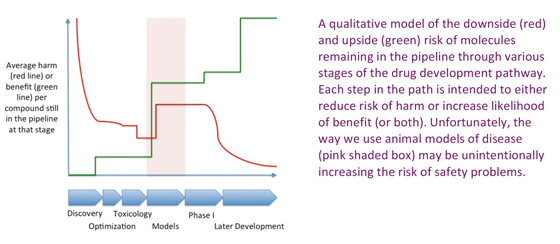
During early screening, things change rapidly. Assay such a cell viability screen out many of the ultra-nasty compounds, while screens for a particular molecular interaction thought to be important for efficacy in a particular indication raise the likelihood that the positive hits might offer benefit. Lead optimization, with a focus on pharmaceutical properties might reduce the risk a bit further by eliminating compounds with more subtle toxic effects, but doesn’t really move the needle in terms of likelihood of eventual efficacy in the clinic.
Once the compounds go into animals, further toxic effects not revealed by sophisticated in vitro screening and in silico modeling will likely be revealed, providing a step change in average safety, again without materially affecting likelihood of efficacy.
But the really interesting thing is what happens when compounds are put through non-clinical efficacy models (the region shaded pink in the figure). The objective is to pick compounds that are likely to have real efficacy against the disease in question, and a lot of time and effort goes in to considering the positive and negative predictive value of various animal models for any given indication. A compound that shows efficacy in a well-validated animal model is suddenly much more likely to eventually show benefit in the modeled disease – hence the big step up in the green line.
You might think such selection on the basis of efficacy would have no effect on the likelihood of harm (after all, why should examining effects in an in vivo model of a disease tell you more about the potential for harm than carefully designed toxicology studies in the same species?). So the red line should be flat – yes? No. Or at least I argue that its not – instead there is a significant INCREASE in risk. It is this proposed increase that lies at the centre of the thesis Im putting forward in this article.
The impact of selection during clinical development is also shown, and is probably fairly uncontentious: risk is largely eliminated early (mostly in Phase I), while eventual proof of efficacy mostly occurs late, just before approval. The products that eventually reach the pharmacopoeia are almost all effective (to some degree) but carry a small residual risk that is difficult, and increasingly expensive, to eliminate entirely.
If the accepted wisdom (and indeed the whole purpose of the drug development pathway) is to elicit a monotonic decline in average risk coincident with a monotonic increase in chance of efficacy, why have I postulated increased risk with increased chance of benefit during animal model screening?
“Like a distorting mirror at a fairground attraction, an animal model is a twisted reflection of a Phase III study – a contraction of time and space. But a contraction that comes at a price”
Because animal models are usually designed to be much more rapid and aggressive than the disease they are modeling. It stands to reason that they should be that way: they are meant to be a quick and easy way to look for efficacy. Typical experiments might have half a dozen animals per group developing a disease, such as rheumatoid arthritis, over a few weeks. Contrast that with even a fairly early Phase II trial with the best part of a hundred subjects and three to six months of treatment, let alone a full phase III study design in hundreds of patients lasting a year or more. The idea is to predict the outcome of the latter trial (which is vast and expensive) from the outcome of the former animal study (which is, relatively, cheap and quick).
Like a distorting mirror at the fairground, the animal model is a twisted reflection of the Phase III study. And this contraction in size and duration is essential: without the ability to put dozens (or industry-wide, thousands) of different promising compounds through such an efficacy screen it would be impossible to select compounds with an adequate chance of showing efficacy in the expensive clinical studies.
But there is a price to pay for the contraction of time and scale. Not just one price but two. The first price, widely recognized and willingly paid, is that the animal model may not be completely predictive of outcome in the clinic. After all, it’s a rat and its not the “real” disease. You cant expect perfection, but you can stack the odds by looking in several models with differing positive and negative predictive powers. Deciding what constitutes a “developable” efficacy profile from such animal model data is an art akin to reading tea-leaves, but it is a closely guarded proprietary skill of any self-respecting pharma company, which differs from company to company, and is no doubt regarded locally in each company as better than the rest.
“Getting a positive result in a rapidly progressing, aggressive animal model is a very high hurdle – requiring strong biological activity. And strong biological activity implies higher risk of side-effects.”
The second price, by contrast, has largely slipped under the radar. Getting a positive result in an aggressive (rapidly progressing) model of disease in just a few animals is a very high hurdle. You have to have a strong biological activity of some kind. A subtle, slowly acting nudge that, over time, builds into a cure has hardly a chance of hitting this particular jackpot. And having such a strong biological activity usually means you have to hit crucial pathways, orchestrating major in vivo responses, which, in turn, means a higher likelihood of side-effects or those dreaded low frequency, high impact toxic events. The analogy is simple: today’s animal models require you to throw a boulder into the pond – with waves fanning out into multiple related systems, rather than targeting the right pressure point with a small pebble that strikes hard where it hits but leaves little more than a gentle ripple propagating away from the epicenter. The gentle but sustained pressure that might be the ideal treatment for the human disease has an imperceptible effect on the raging disease processes in the animal models.
The impact of this selection criterion on the pharmacopeia is clearly illustrated in rheumatoid arthritis: steroids are a great example of the boulder thrown in the pond, while anti-TNFs (the modern pharmaceutical industry’s major contribution to RA treatment) are typified by their Lazarus effect – they have the powerful effect in hours or days that’s need to be shine out in the contracted temporal and spatial dimensions of animal model world. By contrast, most RA patients even today are actually treated with methotrexate – a throw-back to the days before molecular screening and animal models. Methotrexate takes weeks or months to have an effect, but for many patients it does have real clinical benefit over many years. Yet methotrexate has little or no effect on the animal models that dominate drug discovery in RA today, such as collagen-induced arthritis used in treatment model (a model that illustrates very nicely the concept of a quickly progressing, aggressive disease that phenocopies the joint histology seen in human RA, but bears no resemblance to the natural history or time course of the disease).
Of course, methotrexate is not a drug to hold up as a paradigm for a modified drug discovery framework – it’s side-effects make it difficult to use in the clinic and difficult to tolerate for the patient. But the moral of this story is that a modern methotrexate equivalent, free from the side-effects, would be discarded in favour of apparently stronger and more effective medicines. Medicine that, more likely than not, would eventually fail to reach the market due to the risk of side-effects.
In short, the increasing weight put on animal models of efficacy in modern drug development has subtly tipped the balance in favour of progressing agents that carry a higher risk of eventually losing out to the current fetish for zero risk drug products (whether they are culled by the regulator, by their owners or, most painfully of all, by the physicians who fear to prescribe them after approval).
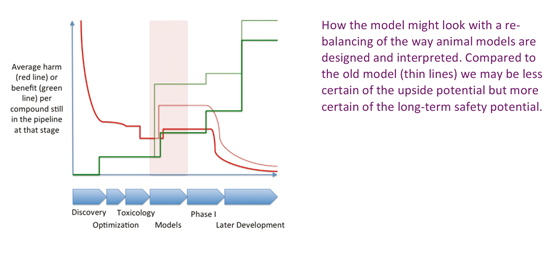
We need animal models of efficacy, just as we need animal models of toxicology. Without their contraction of time and study size, the pipeline for late stage development would dry up almost overnight. But perhaps we have got the balance wrong: for years, we have optimized for quicker, more aggressive models that still predict efficacy because that optimizes the green line in the figure – we can put more molecules through a quicker, cheaper model after all. Yet by turning up the degree of contraction of time and reduction of resource, the hidden penalty of demanding higher and higher levels of biological impact to see a positive outcome is leading to the loss of promising candidates that act more slowly or less aggressively, and which in the long game, may be the only classes of molecules that can meet modern society’s demands for zero risk pharmaceuticals.
The basic, and perhaps surprising, message is simple enough: meeting modern demands for ever improving safety as a commercially viable proposition may require new focus on our animal models of efficacy. Counter-intuitive as it seems, this may be the source of much of our current inefficiency in clinical development. In years to come, we may look back and laugh at the days in the first years of the 21st Century when it was considered the norm to invest in 3 month long GLP toxicology studies before starting a Phase II programme, but still be relying on results from a two week treatment of an aggressive animal model to select for efficacy. If you shoot with a blunderbuss, after all, you are most likely to hit an elephant.
RxCelerate Ltd is an outsourced drug development platform based near Cambridge, UK. We specialize in delivering an entire road map of drug development services from discovery and medicinal chemistry through to formal preclinical development and clinical up to Phase IIa. In the last five years, we have witnessed dramatic changes in the drug development …